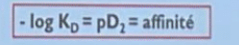Méthode de liaison binding
= permet de déterminer l'affinité du ligand pour le récepteur et d'obtenir des données comme la KD (constante de dissociation) et Bmax (nb total de récepteurs)
Méthode de saturation ou de Scatchard
-> comptage de la radioactivité permet de déterminer la quantité de ligand fixe et non fixé
Matériel
- différents tubes à essai contenant des récepteurs fixés à la membrane à la même concentration
- un blanc = témoin
- ligands radio-marqués (molécules qu'on veut tester) tester à différentes concentrations (croissantes en ligand marqué)
- on mélange les ligands marqueurs avec les récepteurs
- rinçage = on retire le ligand qui n'a pas été fixé
- filtrage
- détermination des différentes concentrations ligands-marqués liés et non liés
- ce qui est resté sur le filtre = lié au récepteur
- ce qui passe à travers le filtre = ligand libre
- représentation graphique ligand-libre/ligand lié -> détermination de l'affinité
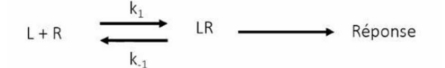
-> fixation du ligand sur le récepteur entraîne la formation d'un complexe LR = on filtre puis on compte
Protocole
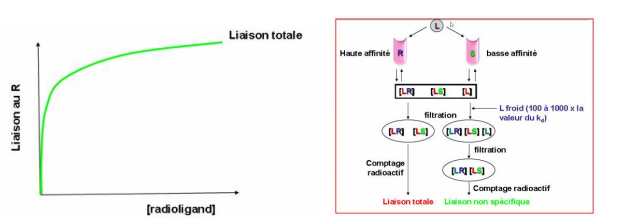
-> sur une autre série de tube : on refait la même chose avce un ligand non spécifique et un libre
- on ajoute un ligand froid non radioactif = va beaucoup plus rapidement se fixer sur le site spécifique
- = liaison non spécifique (à revoir avec ronéo)
- ligand froid remplace dans la majorité des cas le ligand radioactif sur le récepteur = compétition
- sur les sites non spécifiques = pas de compétition = le liaison non radioactif n'est pas remplacé = liaison non spécifique avec le ligand radioactif
- on mesure la concentration = radioactivité = ligand radiomarqué de manière non spécifique
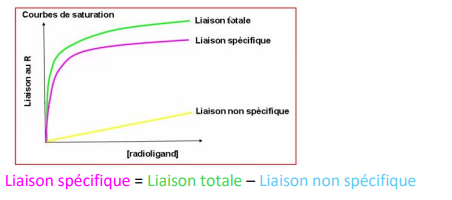
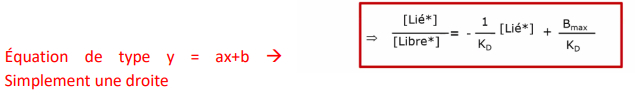
liaison spécifique permet de déterminer précisément l'affinité entre le ligand et le récepteur
Modification de l'équation KD
Interaction médicament-récepteur
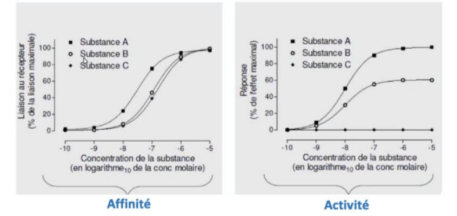
-> affinité :
- substance A va se fixer sur la première sur les récepteurs, elle aura donc une affinité plus élevée que les substances B et C pour ces récepteurs en question
- de + il y a besoin de moins de substance A que de B pour arriver à 50% de l'effet max = + puissante
-> activité :
- substance A va avoir une activité plus importante que les substances B et C
- substance B aura donc une activité par rapport à la substance A mais une activité plus importante que la substance C
- substance C n'entraîne aucun effet, elle n'a donc aucune activité - elle empêchera seulement A et B de se lier sur les récepteurs = antagoniste
-> plus le KD est faible -> plus l'affinité est élevée
Sélectivité
-> ex de la noradrénaline
-> ligand = interaction avec plusieurs sous-types de récepteurs
- pas de spécificité absolue
- action de manière relativement sélective
- sélectivité = dépendante de la liaison physicochimique du médicament aux récepteurs
- ex : les neuroleptiques - antipsychotiques
-> effet des neurlopetiques
- chlorpromazine : {1 = 5HT2 > D2 > D1
- cela signifie que la chlorpromazine a une forte affinité pour les récepteurs α1-adrénergique et 5HT2 plus que pour les récepteurs dopaminergiques D2 et D1
- halopéridol : D2 > D1 = D4 {1 > 5HT2
- affinité pour les récepteurs dopaminergiques D2 est prédominante
- clozapine : D4 = {1 > 5HT2 > D2 = D1
- affinité préférentielle pour les récepteurs D4 et α1 avant les 5HT2 et enfin les D2/D1
Activité intrinsèque
= désigne les effets de différents agonistes sur un même récepteur
- effet différent de l'effet max EmA
-> ex :
- effet contractant sur l'intestin grêle
Définitions
- affinité : aptitude d'un ligand à se fixer sur le récepteur = puissance
- plus la concentration qui va donner un effet sera faible + la molécule est puissante
- activité intrinsèque : capacité d'un ligand à entraîner une réponse quantifiable après fixation = efficacité
Pour un effet intermédiaire :
-> différentes activités intrinsèques possibles :
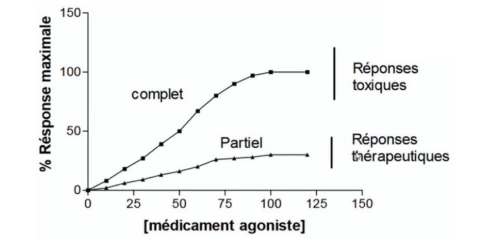
- agoniste entier : α = 1
- agoniste partiel : 0 < α < 1
- = va entraîner le même effet qu'un agoniste entier mais d'une manière moins importante
- antagoniste : α < 0
- = activité intrinsèque est égale à 0 car elle n'entraîne pas d'effet - il se fixe sur le récepteur mais n'agit pas
Agonistes partiels
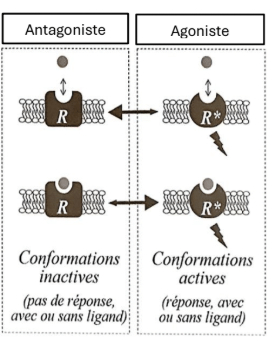
-> modèle à 2 états du récepteur
-> oscille entre 2 états :
-> ligand - en fonction de son activité intrinsèque - va soit stabiliser une forme active = effet plus durable
= agoniste stabilise l'état actif et antagoniste stabilise une activité intrinsèque
Thérapeutique
-> ttt de la dépendance
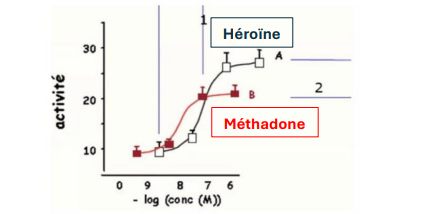
- méthadone pour l'héroïne
- = + puissante que l'héroïne = n'entraîne pas l'effet de l'héroïne = pas de manque = pas de flash
- -> agoniste partiel = comble les récepteurs
-> on traite le syndrome de sevrage -> on fait de la réduction de risque mais en réalité ne se soigne pas
-> cardiologie
- 2 formes d'états bétabloquants
- sans ASI = ce sont des vrais antagonistes - se fixent sur les récepteurs β et empêche leurs effets
- ex : propranolol
- avec ASI : font la même chose que les agonistes entiers mais à un niveau moindre - agonistes partiels
- ex : acébutolol
-> β avec ASI :
- font la même chose que l'agoniste du système sympathique en stimulant les récepteurs β1 mais à un niveau moindre
- effet β mais ne vont pas trop loin contrairement aux autres antagonistes (= ne diminuent pas complètement le rythme cardiaque)
- = évitent ainsi un effet rebond du rythme cardiaque + bradycardie trop importante
Effets
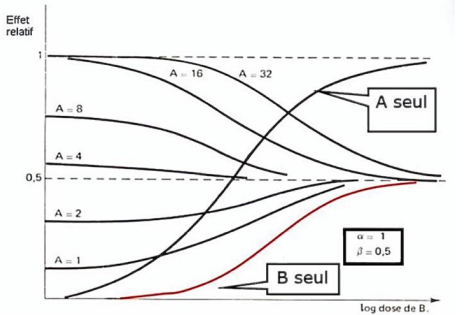
-> courbe "A seul" est un agoniste entier = par ex noradrénaline qui stimule le rythme cardiaque
-> courbe "B seul" est un bêtabloquant avec une activité partielle
- quand on augmente la concentration en noradrénaline seul on obtient un effet max
- quand on augmente la concentration de B seul - on obtient un effet mais plus faible (50% de moins)
- B va donc produire le même effet que A = activité sympathomimétique - mais de façon moins importante
Agoniste inverse = antagoniste négatif
-> 2 effets différents en fonction du niveau basale :
- antagoniste
- agoniste inverse
Antagonisme
= opposition partielle ou totale d'un PA aux effets d'un autre PA
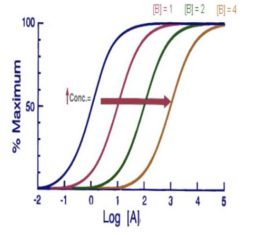
Antagonisme compétitif réversible
-> agoniste A, d'affinité 1/KA et d'activité intrinsèque alpha > 0
-> antagoniste B d'affinité 1/KB et d'activité intrinsèque bêta = 0
-> interaction réversible de A et B avec le récepteur
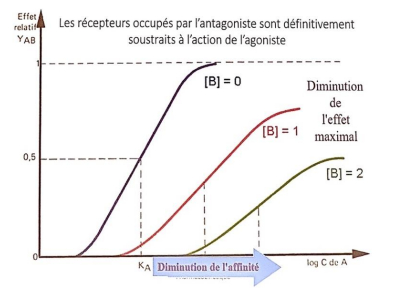
-> la présence de B diminue l'effet de A
- on aura un déplacement de la courbe de A vers la droite = diminution d'affinité mais activité équivalente : + la concentration de B est importante + on doit augmenter la concentration de A pour conserver un effet max
- blocage est surmonté car B est un antagoniste de B et retrouve un effet maximal
Affinité de l'antagoniste
-> constante pA2
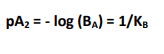
= logarithme de la concentration en antagoniste qui nécessite le doublement de la concentration en agoniste pour obtenir le même effet
Antagonisme compétitif irréversible
= diminution de l'effet et de l'affinité
- la courbe se déplace vers la droite et redescend
-> une fois que le récepteur est occupé = irréversible - on ne pourra pas le déplacer
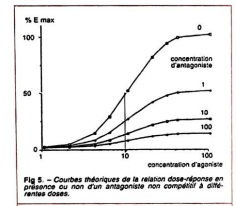
Antagonisme non compétitif
-> hausse de concentration de l'antagoniste
-> pas de compétition mais n'entraîne pas de diminution de l'affinité
= diminution de l'effet max
Antagonisme fonctionnel
= 2 agonistes modifiant un paramètre biologique en sens opposés
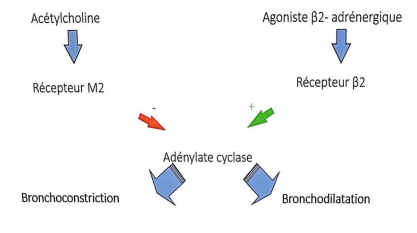
acétylcholine = système parasympathique
-> ex :
- effet de l’acétylcholine sur son récepteur M2 va diminuer l’effet de l’adénylate cyclase = diminue les concentrations en AMPc, donc il entraîne une bronchoconstriction. - À l’inverse, un agoniste β2-adrénergique (comme la terbutaline), va se fixer sur les récepteurs β2 et augmenter l’effet de l’adénylate cyclase, donc augmenter les concentrations en AMPc, donc il entraîne une bronchodilatation.
Antagonisme par interaction pendant la phase pharmacocinétique
-> antagoniste intervient en diminuant la biodisponibilité de l'agoniste pour son récepteur et non plus sur le récepteur lui-même
- diminution de la résorption de l'agoniste
- accélération du métabolisme de l'agonsite
- augmentation de l'élimination urinaire de l'agoniste
Inversion d'action
-> modification de l'effet avec la dose
-> dépresseurs du SNC
- niveau d'excitabilité : peut aller des convulsions jusqu'au coma
- équilibre entre l'activité de systèmes excitateurs et inhibiteurs
-> vigilance : conséquences d'un antagoniste de la formation réticulée activatrice et de la formation réticulée inhibitrice
-> à faible dose : levée de l'inhibition
- les anesthésiques volatils
- certains barbituriques
- éthanol
- induisent une phase d'excitation
- = action préférentielle de ces dépresseurs sur les systèmes inhibiteurs
- augmentation de l'activité des centres excitateurs
-> à forte dose :
- agissent sur l'ensemble du SNC
- = dépression de l'ensemble des centres excitateurs et inhibiteurs
Synergie
= interactions entre les substances pharmacologiquement actives ayant des effets agonistes
- = implique que des agonistes agissent ensemble pouvant aboutir soit à un effet total supérieur à la somme des effets individuels - soit à une diminution de l'effet dans certains cas spécifiques
Potentialisation
= synergie imparfaite où l'effet global est supérieur à celui des substances prisés séparément mais sans être strictement additif
Synergie directe
= interaction/effet de 2 médicaments
-> récepteurs spécifiques différentes
- = interaction à l'origine d'une action pharmacodynamique
- ex : anesthésie
- anesthésiques : diminution de l'activité musculaire incomplète
- + curares
- = augmentation du relâchement musculaire
Synergie indirecte
= médicament B supprime une fonction antagoniste de l'effet de A
-> ex :
- effet de l'administration conjointe de sympathomimétiques et de parasympatholytiques
- au niveau cardiaque :
- les sympathomimétiques
- récepteurs bêta = effet chronotrope et inotrope positif
- les parasympatholytiques
- récepteurs M = diminuent l'effet chronotrope et inotrope négatif de l'acétylcholine
Interaction dans la phase pharmacocinétique
-> augmentation du degré ou de la vitesse de résorption d'un médicament A par une substance B (excipient)
-> réduction de l'élimination de A par B
- utilisé dans le ttt du VIH par ex
-> déplacement de la fixation de A aux protéines plasmatiques par B
- important pour les médicaments très liés aux protéines plasmatiques
- si on 50% de fixation (forme de réserve dans le compartiments sanguins) -> 50% libre (diffuse dans les tissus) = qui va entraîne l'effet
- déplacement -> pour avoir un effet sur l'effet = fraction libre multipliée par 2
- si très fortement lié aux protéines plasmatiques 99% = forme libre représente 1% = on peut passer à 2% si déplacement = augment les EI
-> inhibition du métabolisme de A par B
- réduction de l'élimination
- plus utilisés