ADN recombinant provient d'une combinaison entre :
- ADN d'un organisme donneur = ADN à cloner
- ADN d'un vecteur = ADN hôte
-> biotechnologies et santé :
- prévention
- diagnostic
- nouveau médicament = bio médicament
- production de protéine recombinantes :
- hormones
- facteur de croissance
- vaccins recombinants
- facteur de coagulation
- anticorps
Clonage
= isoler et obtenir de nombreuses copies identiques d'un gènes ou d'un fragment de gène
-> conservation parfaite de l'info génétique
- à l'échelle cellulaire = clonage cellulaire
- à l'échelle de l'organisme = clonage reproductif
-> clone : grand nombre de cellules ou de molécules identiques et provenant d'un seul ancêtre commun
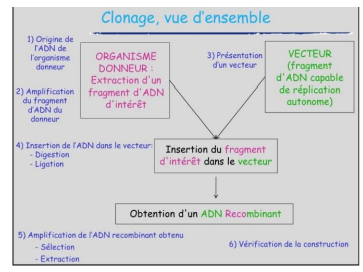
Principe générale
1-Organisme donneur d'où on extrait le fragment d'ADN d'intérêt
2-Le vecteur qui permet d'amplifier le gène d'intérêt
3-On insert le fragment d'intérêt dans le vecteur pour obtenir un ADN recombinant
4-Amplification de l'ADN et expression de la protéine correspondant à l'ADN si on utilise le bon vecteur
Origine de l'ADN de l'organisme donneur
-> 2 possibilités
- ADN génomique = ADN représentant l'ensemble du génome codant et non codant
- extrait à partir d'une culture cellulaire
- ADN complémentaire = ADN ne contenant que les séquences codants (plus d'introns) des gènes
- ADN obtenu par RT des ARNm

Rappel transcriptase inverse
-> ARNm a une queue polyA en 3' qui permet d'utiliser un oligonucléotide
- 1ère étape : hybridation avec l'oligonucléotide complémentaire de la queue polyA
- 2ème : élongation par la reverse transcriptase qui permet d'obtenir un brin d'ADN complémentaire
- 3ème : synthèse du second brin d'ADN complémentaire par une ADN polymérase
-> ARNm est éliminé par un ttt à la RNase
-> synthèse du second brin d'ADNc assuré par une ADN polymérase
= on part de l'ARNm puis on obtient un ADNc dont les séquences non codantes sont éliminés
Amplification du fragment d'ADN d'intérêt
-> besoin de :
- 2 amorces
- 4 dNTP
- Taq polymérase
- ADN à amplifier
- Mg
- Tampon salon
- amplification exponentielle
-> PCR en 3 étapes :
- dénaturation
- hybridation
- élongation
-> amorce dans la zone amplifiée
-> environ 30cycles et à chaque cycle on double la quantité d'ADN l'amplification est exponentielle
-> après 28 cycles on a 228 molécules d'ADN
-> utilisation de l'électrophorèse en gel d'agarose pour voir si la PCR a fonctionné + BET
-> si la PCR fonctionne = 1 seul bande - les fragments ont la même taille
- on utilise un marqueur de taille en pb
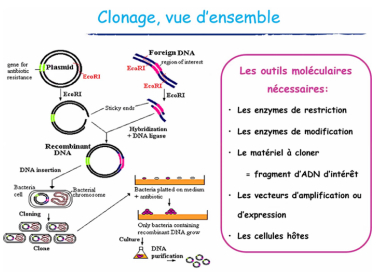
-> clonage repose sur l'insertion d'un fragment d'ADN exogène d'un vecteur = permet ensuite d'exprimer l'ADN dans les cellules hôtes
Présentation d'un vecteur de clonage
-> ADN ne peux pas pénétrer librement dans une cellule = un vecteur est un transporteur permettant l'introduction et la multiplication d'un séquence d'ADN dans une cellule hôte
-> grande diversité
- cosmide
- bactériophage
- chromosomes artificiels de levures (YAC) et de bactéries (BAC)
-> vecteurs utilisés en génie génétique ont souvent une origine naturelle bactérienne mais ont été modifiés
-> différents types
- vecteurs d'amplification
- amplifie l'ADN cloné = nombreuses copies possibles
- vecteurs d'expression
- amplifient l'ADN en se multipliant mais sont capables de transcrire et traduire un ADNc = synthèse de protéine recombinante correspondante
-> vecteur ne se réplique pas seul = il lui faut une cellule hôte
-> à chaque vecteur = un hôte cellulaire
- cellule procaryote
- cellule eucaryote
= hôte cellulaire choisi en fonction des buts initiaux
Propriété d'un vecteur de clonage
-> capacité de réplication autonome dans une cellule hôte donnée
- réplication est bidirectionnelle = on obtient un plasmide fils qui assure une stabilité
- = réplication épisomale indépendante de celle de l'ADN de la cellule hôte pour augmenter la stabilité du système
-> possession d'un site de clonage multiple pour l'insertion d'un fragment d'ADN
- = différents sites uniques de restriction donc les sites ou le vecteur peut être ouvert en utilisant la bonne enzyme de restriction -> manipulable facilement
- permet de choisir une enzyme de restriction pour manipuler facilement le vecteur en l'ouvrant puisqu'un plasmide est circulaire et bicaténaire
-> insertion d'un fragment d'ADN plus ou moins grand à l'intérieur du vecteur influence le choix du vecteur
-> présence fréquente d'un marqueur de sélection
- = gène de résistance à un ATB, marqueur de sélection métabolique qui permet de sélectionner des bactéries
Exemple du plasmide, vecteur d'expression
-> plasmide
- petite molécule extra chromosomique d'ADN
- double brin, circulaire
- entre 2 et 5 kb
- capable de se répliquer indépendamment du chromosome bactérien
- peut être transféré d'une cellule à une autre
Un plasmide adopte différentes conformations
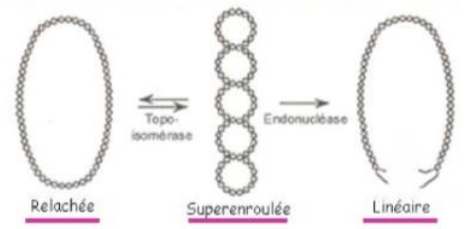
-> forme relâchée : plasmide sous forme circulaire, sans surenroulement
- topoisomérases permettent de passer d'une forme relâchée à super enroulée
-> forme superenroulée : forme intermédiaire
- endonucléase clive le plasmide superenroulée pour donner la forme linéaire
-> forme linéaire : qui a été clivé par une enzyme de restriction
Insertion de l'ADN d'intérêt dans le vecteur
-> en 2 étapes
- digestion enzymatique
- ligation
-> mécanisme d'action des enzymes de restriction
- enzyme de restriction est mise en présence d'ADN double brin
- enzyme de restriction se lie à une séquence spécifique
- coupe les 2 brins d'ADN à des endroits précis
- se libère et l'ADN est fragmenté en 2
-> 2 types de coupures générées
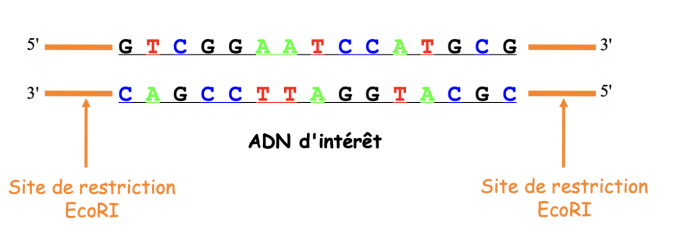
- bouts cohésifs : enzyme ne clive pas au même niveau des 2 brins (Eco R1)
- bouts francs : coupure au même niveau, au milieu des 2 brins d'ADN (Sma 1)
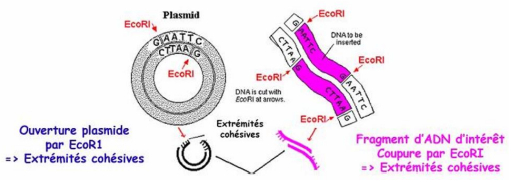
Digestion du fragment d'ADN et du vecteur
-> plasmide bicaténaire circulaire et fragment d'ADN à amplifier (rose)
- on choisit l'enzyme de restriction Eco R1, le plasmide est ouvert par digestion enzymatique
- cette enzyme génère des extrémités cohésives
- en parallèle on coupe le fragment d'ADN d'intérêt avec Eco R1 qui génère également des extrémités cohésives
-> digestion enzymatique : doit se faire dans un tampon adapté à l'enzyme et à la température optimale de l'enzyme, elle dure souvent 2h
-> plasmide ouvert + ADN après digestion enzymatique mais comment insérer dans le vecteur ?
- quand on utilise une enzyme de restriction le fragment d'ADN va pouvoir s'insérer dans 2 sens différent du vecteur
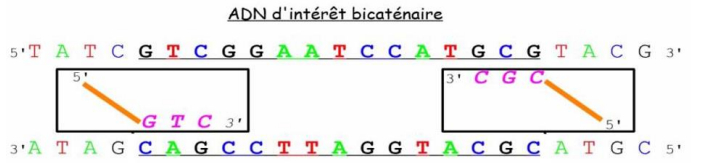
-> la séquence d'ADN peut s'orienter dans 2 sens différents
- se fait au hasard
- peut être gênant si on veut synthétiser la prot recombinante de l'ADN que l'on a cloner mais si on veut amplifier une séquence ça ne pose pas de problème de ne pas maîtriser le sens d'insertion
Problématique
- quand on utilise qu'une seule enzyme de restriction le problème = on ne maîtrise pas le sens d'insertion de ce fragment d'ADN
- = 2 sens d'orientations possibles du fragment d'ADN à l'intérieur du génome ce qui peut être gênant ou non
- pas gênant dans le cas où notre objectif est d'amplifier un fragment d'ADN
- mais gênant quand on veut exprimer une prot dans ce cas car le sens est très important
-> les ajouter aux extrémités de l'ADN à cloner
-> 1ère possibilité :
- = on effectuer une PCR classique mais les amorces ne seront pas les mêmes que d'habitue
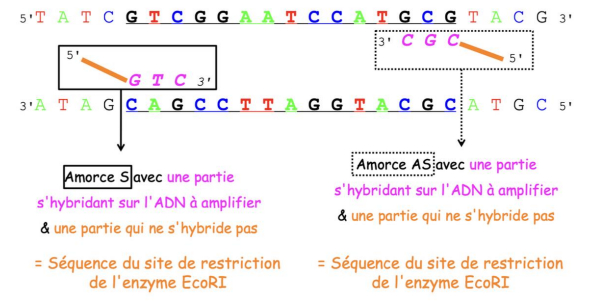
= nous aurons d'abord une Amorce S avec une partie s'hybridant sur l'ADN à amplifier et une partie qui ne s'hybride pas
-> de la même manière nous aurons une Amorce AS avec une partie qui s'hybride sur l'ADN à amplifier et une partie qui ne s'hybride pas
= on se retrouve avec notre ADN d'intérêt qui a été amplifié de part et d'autre de nos sites de restriction qui vont permettre d'envisager le clonage
-> clonage possible
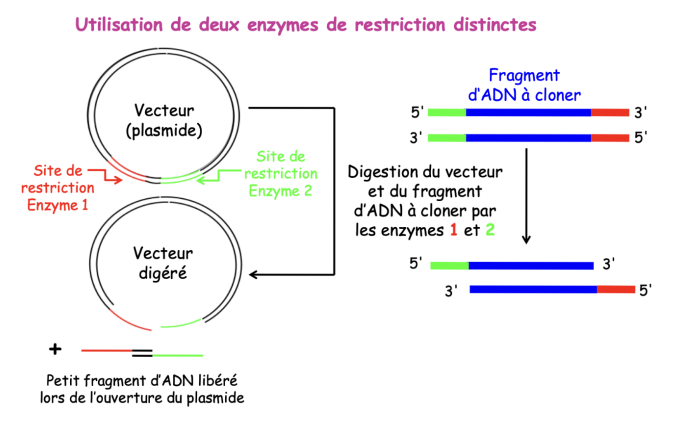
-> 2ème possibilité :
- = utilisation de 2 enzymes de restrictions distinctes
- intérêts :
- insertion orientée de l'ADN à cloner dans le vecteur
- limitation de la relégation du vecteur sur lui-même
-> on effectue d'abord une double digestion enzymatique = une seule possibilité d'insertion = on maîtrise complètement le sens d'insertion du vecteur
= on privilégie cette méthode
= lorsqu'il y a deux enzymes de restrictions différentes = une seule possibilité d'insertion du fragment d'ADN dans le vecteur
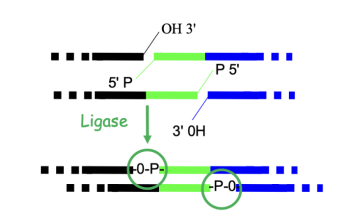
Ligation du fragment d'ADN dans le vecteur
-> ligase : enzyme capable de former des liaisons covalentes
- en présence d'ATP = formation de liaisons phosphodiester entre 5'P et 3'OH de 2 brins adjacents
-> fragment d'ADN d'intérêt + vecteur + liaisons phosphodiester = plasmide recombiné
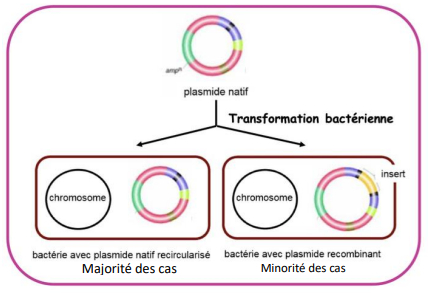
Amplification de l'ADN recombinant
-> faire pénétrer une molécule d'ADN dans une cellule pour laquelle il est étranger = transformation de la cellule hôte -> amplification du vecteur par la machinerie de la cellule hôte
-> avant toute transformation, les bactéries sont rendues compétentes
- leur état physiologique doit garantir une efficacité max de transformation
-> action du chlorure de calcium (CaCl2)
- ions Ca2+ forment des nanopores réversibles dans la membrane bactérienne -> paroi fragilisée -> entrée d'ADN étranger facilitée
Transformation bactérienne
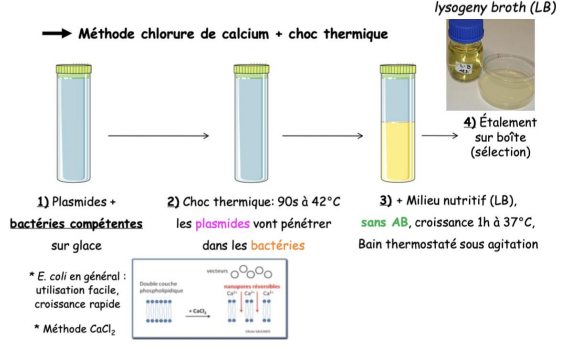
- 1- plasmide + bactéries compétentes
- choc thermique : 90 sec à 42°C -> pour que les plasmides pénètrent dans la bactérie
- ajout du milieu nutritif sans ATB 1h sous 37°C dans un bain thermostaté sous agitation pour exprimer la résistance aux ATB dans la bactérie
- étalement d'une petite partie du milieu des bactéries puis ajout de l'ATB d'intérêt pour ne sélectionner que les bactéries qui ont ingéré le vecteur
-> pourquoi exprimer la résistance plasmidique
- pour ne sélectionner que les bactéries qui ont intégré le vecteur pas recombiné
Sélection des bactéries transformées
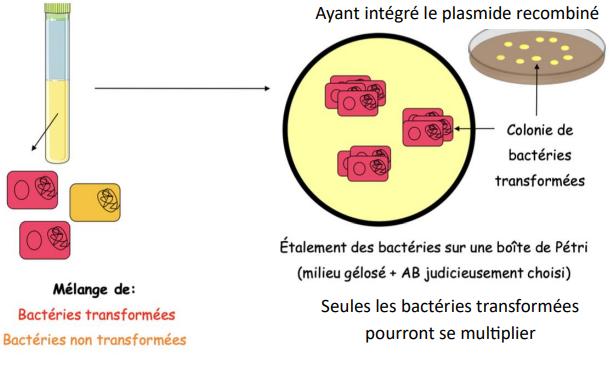
-> si on considère qu'une seule colonie = elles auront toutes le même génotype
Amplification du plasmide recombiné
-> ensemencement d'une colonie de bactérie transformée dans le milieu liquide + ATB
-> une nuit à 37°C sous agitation
= multiplication de bactérie et amplification du nombre de plasmide recombiné
-> risque d'utiliser qu'un seul erlenmeyer est d'obtenir un plasmide vide pour éviter cela = conseillé d'en faire 10 en parallèle en faisant 10 erlen et en choisissant plusieurs colonies dans chaque erlen
-> pour bien choisir la colonie il faut repérer celles qui sont grosses et + éloignées les unes entre elles
-> objectif = extraire le plasmide
Extraction du plasmide recombiné
-> bactéries sont lysées par un détergent en milieu alcalin
- libération de l'ADN génomique et plasmidique
- ADN génomique et les protéines sont précipitées par de l'acétate de sodium
- précipité est séparé par centrifugation
- surnageant contient de l'ADN plasmidique
- ADN plas est alors précipité, lavé puis redissout dans un tampon adéquat
-> au début 10 relents puis à ce stade = 8
Vérification de la construction obtenue
-> vérification de l'ADN obtenu
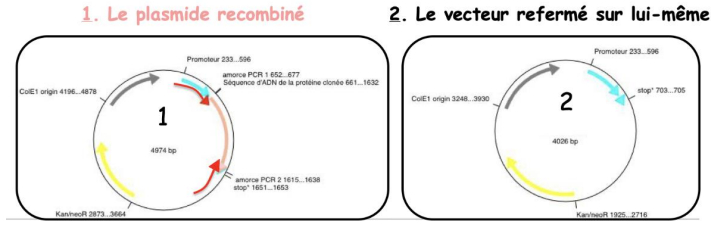
-> pb : bactéries poussent sur le milieu + ATB possèdent un plasmide avec le gène de résistance à l'ATB
- mais ce plasmide contient-il l'insert cloné ?
-> relégation du vecteur sur lui-même possible
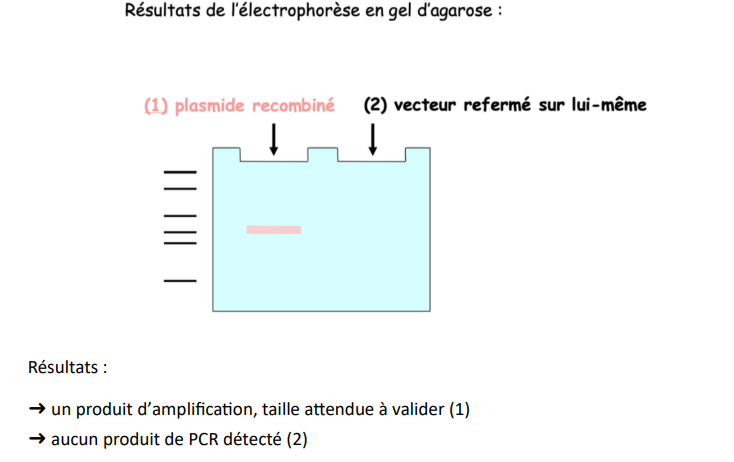
Vérification par PCR
-> bactéries transformées peuvent contenir
-> comment distinguer ces 2 populations
- PCR avec les amorces ayant servi à obtenir de l'insert et observation des produits de PCR sur gel d'agarose
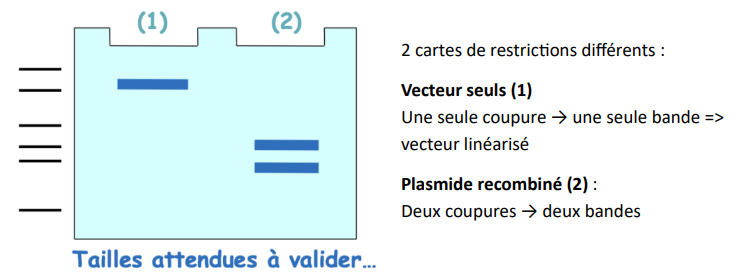
Vérification rapide par restriction
-> digestion enzymatique par un enzyme ayant
- un site de R dans l'insert
- un site de R dans le vecteur
-vecteur qui fait 4000pb = on attend une bande à 4000pb
-si on le fait migrer tel quel sans digestion on va obtenir différentes bandes avec différents degrés de surenroulement
= vecteur recombinant sera coupé au niveau de l'insert donc on attend d'obtenir 2 fragments de tailles différentes
Analyse des produits de restriction par électrophorèse sur gel d'agarose
Vérification par séquençage : méthode de Sanger
-> pb : si par ex 1 seul vecteur = une coupure = une bande
- on va re prélever des colonies différentes des premières si possible bien isolées les uns des autres et on recommence le processus
conclusion :
- obtention d'un ADN recombinant amplifié